











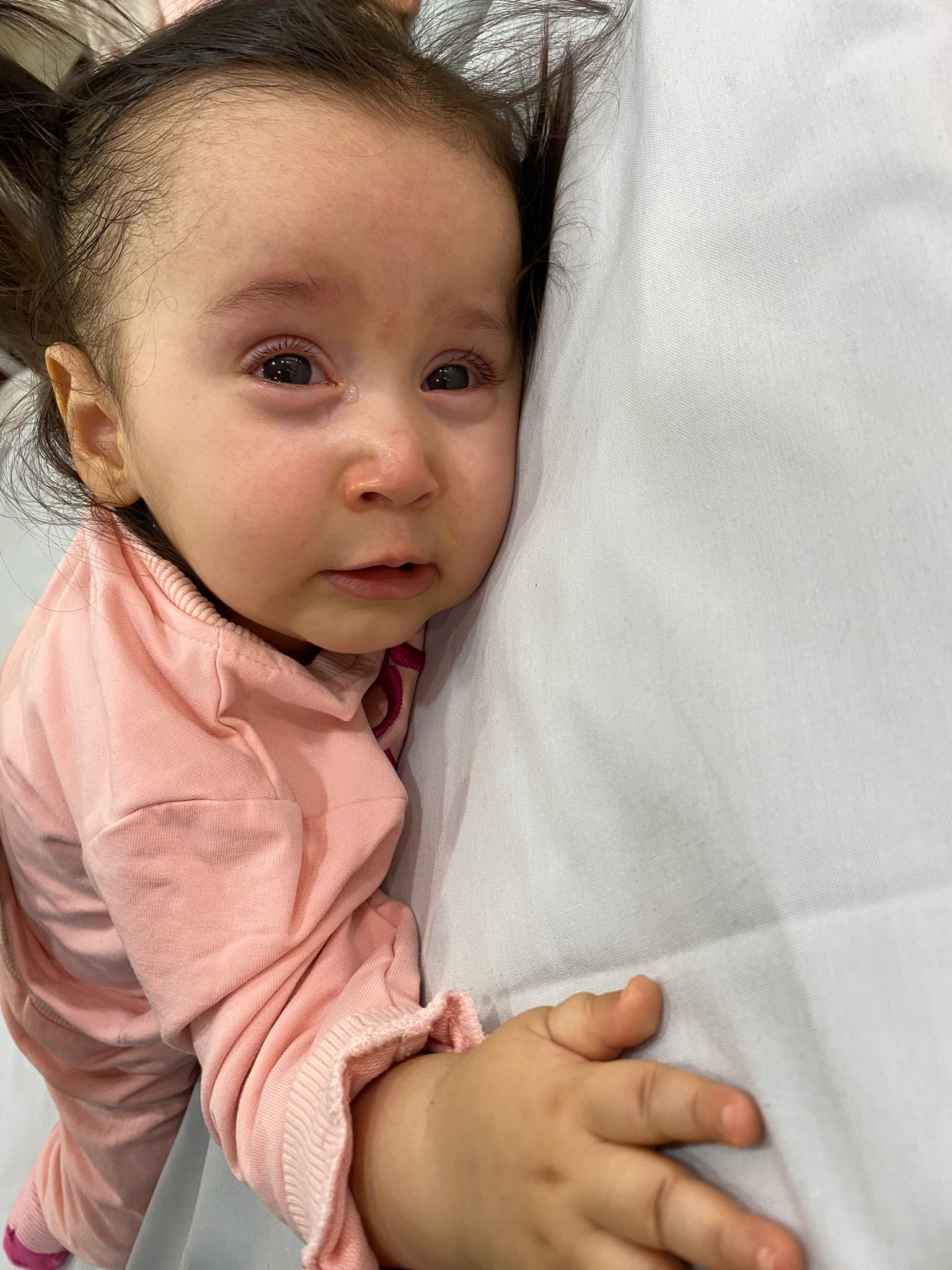
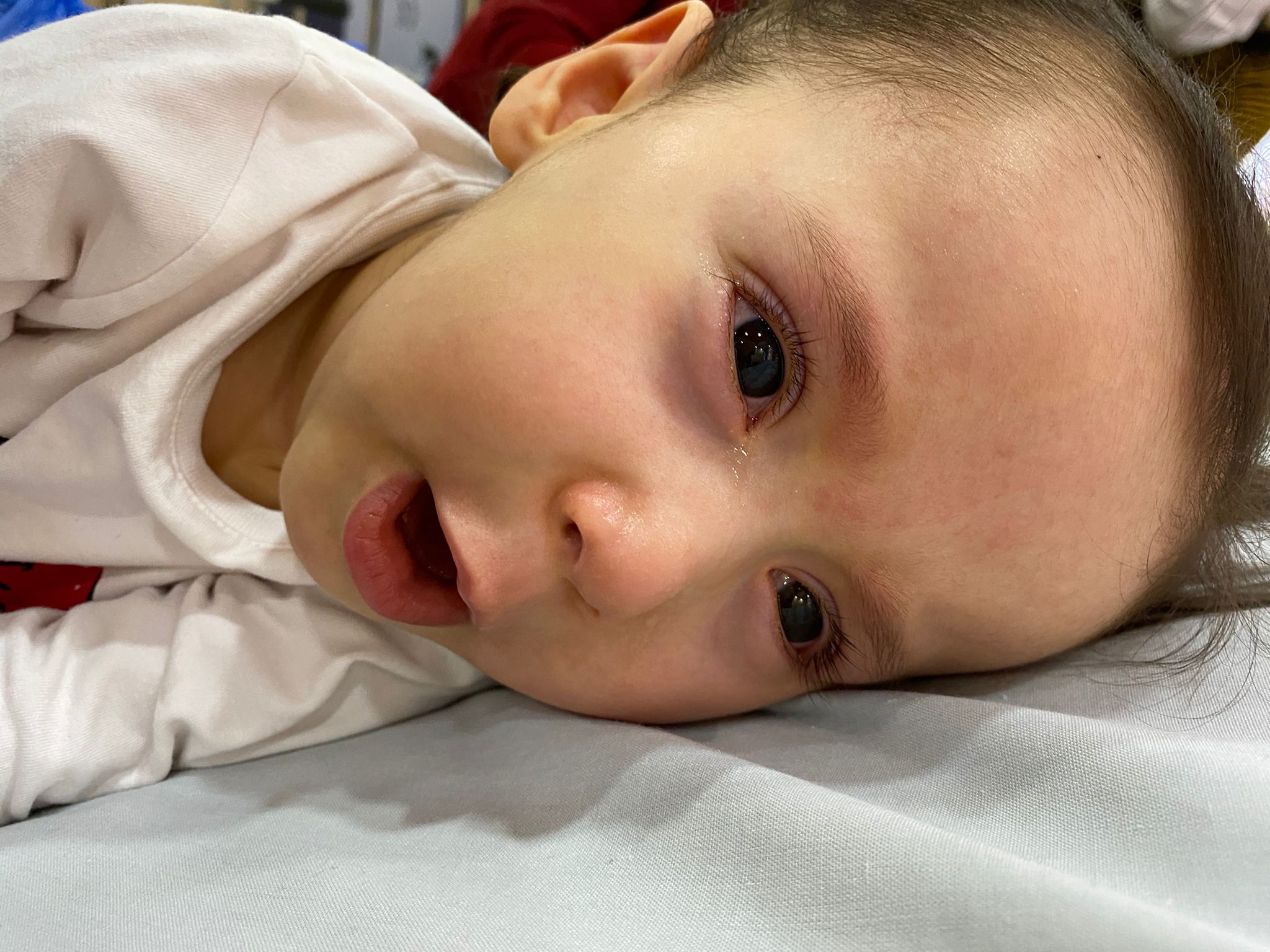
Tüm Türkiye’de sayıları binleri geçen SMA hastalarının yardım çığlıkları devam ediyor. Bir yardım çığlığı ise Şanlıurfa’da yükseldi. SMA Tip 1 hatası Alara Şahbaz’ın iyileşmesi için ailesi yardım severlerden destek bekliyor.
Konu ile ilgili açıklamalarda bulunan anne Güler Şahbaz, maddi durumlarının ancak kendilerine yetebilecek düzeyde olduğu kızlarının tedavisini sağlayacak maddi durumlarının olmadığını belirterek şu ifadelere yer verdi:
“Merhabalar kızımız Alara Şahbaz, SMA Tip 1 hatası. Ben Güler Şahbaz annesiyim öğretmenim, eşim Ersel ŞAHBAZ mühendis. Kendi hayatımızı idame ettirebilecek güce sahibiz ama kızımızın tedavisini sağlayacak maddi imkana sahip değiliz.
SMA’yı kısaca özetlemek gerekirse; vücudumuzda SMN proteinini üreten SMN1 geninin mutasyona uğrayarak protein üretememesi sonucu oluşan ilerleyici bir kas hastalığıdır. Vücudumuz yeteri kadar protein üretemediği için motor nöron sinir hücrelerimiz beslenemiyor. Bunun sonucunda zamanla kaslarımız zayıflıyor ve atrofi (kas kayıplarına) neden oluyor. Bu hastalığın 4 çeşidi var. SMA TİP1-2-3-4 Alara SMA Tip 1 hastası ve belirtileri en ağır grupta yer alıyor. Normalde bizim çocuklarımız için 2 yaşına kadar yaşar, anı biriktirin denilen çocuklardır. Çünkü hastalık öyle bir şeyki: -Normal çocuklar gibi kafamızı tutamıyor bu yüzden hem oturamıyor hem ayakta duramıyoruz. -Yürümek bizim çocuklarımız için imkânsız.-Tedavi olunmazsa ve hastalık ilerler ise çocuklar zamanla kollarını bacaklarının kafalarını oynatamıyorlar hatta mimiklerini bile yapamıyorlar. -Hastalık daha da ilerleyince kendi başlarına nefes alamıyorlar. Çünkü ilk önce solunum kasları zayıflıyor ve çocuklarımızın boğazına trakeostami açılarak(boğazı delinerek) makine ile nefes almak zorunda kalıyorlar.”
“BİR SONRAKİ DOZU ALABİLİRMYİZİN STRESİNİZ YAŞIYORUZ”
Açıklamasının devamında kızının durumu ile ilgili bilgi veren anne Şahbaz, şunları kaydetti:
“Kızım Alara bu hastalık sebebiyle dönemiyor, emekleyemiyor, desteksiz oturamıyor ve yürüyemiyor. O bizim ilk ve tek çocuğumuz. Alara 10.06.2019’da doğdu. İlk kez 4 aylıkken bacak hareketlerinin yavaşlaması ile hastalığı fark ettik fakat şimdiye kadar ailelerimizde böyle bir hastalık olmadığı için ne olduğunu anlayamadık. Fakat hareketleri giderek yavaşladığı gibi diğer gelişim özelliklerinin hareketlerini de yerine getiremedi. Hemen doktora gittik ve testler yapıldı, 10 aylıkken kesin olarak kızımızın SMA Tip1 Hastası olduğunu öğrendik. Şuan Türkiye’de SGK kapsamında aldığımız Spinraza ilacı var. Bu tedavi beldeki omurilikten sıvı alınıp ilaçla karıştırılarak tekrardan aynı yerden enjekte edilerek uygulanıyor. Bu ilacım 4 dozunu kesin olarak alıyoruz, sonrasında solunum makinesi ve fiziksel hareketleri açısından kendi belirledikleri bir takım kriterleri yerine getirmesi gerekiyor. Bu kriterlerden puanlamaya tabi tutuluyor. Bu ilacı ömür boyu alması gerekiyor ama puan artışı olmazsa ilaç kesiliyor. Biz 5 doz aldık şu anda. Ama her defasında bir sonraki dozu alabilecek miyiz alamayacak mıyız stresini yaşamaktan çok yorulduk.
Ayrıca SMA Hastalarında SMN1 Geninin sıfır (0) kopyası ve bir de bu genin kopyası olan SMN2 geni var. Bu SMN2 Geni ise asıl gen olan SMN2 Geninin sadece %10’u kadar motor nöron üretebiliyor. Spinraza adlı ilaç SMN1 genine müdahale etmiyor, SMN2 geninin verimliliğini artırıyor. Ulaşmaya çalıştığımız tedavi ise Zolgensma Gen Terapisidir. Bu terapide SMA hastalarında bozuk olan SMN1 geni yerine sağlıklı olarak sentezlenen SMN1 geni vektör yardımıyla gönderiliyor. Zolgensma sadece tek bir doz olarak 60 dakikada tamamlanan bir operasyon. Bu ilaç Amerika’nın Gıda ve İlaç Enstitüsü (FDA)’nün verdiği karar ile 2 yaş altı tüm SMA hastalarını kapsıyor.”
“KAMPANYAMIZda YÜZDE 73’E GELDİK”
Tedavinin yapıldığı ülkelerinde ismini söyleyen Güler Şahbaz, yapılacak destekler ile kızının tedavisinin hızlanacağını belirterek şunları söyledi:
“Tedavi Avrupa ülkelerinde , ABD, Dubai ve Japonya’da uygulanıyor. Japonya dışarıdan hasta almıyor. Maalesef bu Zolgensma Gen Tedavisi Türkiye’de uygulanmıyor. İlacın en etkili olduğu zaman 2 yaş altı ve 13,5 kilonun altıdır. Alara 21 aylık ve 9.5 kilo. Biz hastanelere başvurularımızı yaptık. Fiyat ülkeden ülkeye hastaneden hastaneye farklılık gösteriyor. Biz Dubai’den 1.8 milyon Euro fiyat aldık. En uygun teklifi Dubai’den aldığımız İçin orayı tercih ettik.
Biz dünya çapında bir kampanya yürütüyoruz. 6,5 ay oldu kampanyamız. Tedaviyi 2 yaş altında alabilmemiz için biraz daha hızlanmamız gerekiyor. Ne kadar erken alabilirsek hastalık kızımızda daha fazla tahribata sebep olmayacak ve kızımız daha hızlı gelişim gösterecek.
Kendimizin ve Alaramızı sahiplenen hayırseverlerinizle kampanyamızda %73’e geldik. Dubai’den aldığımız hastane teklifi 1.800.000 milyon Euro. Biz bunun 1.314.569 milyon Euro’sunu topladık.
Bizim gücümüz bu tedavi masrafını karşılamaya yetmiyor. Alara’nın da diğer çocuklar gibi oyunlar oynamasını, oturup, yürüyebildiğini ve okullara gidebildiğini görmek istiyoruz. Bu tedavi kas erimesini kalıcı olarak durduruyor. Çocuğumuz tedavi olmazsa onu kaybedeceğiz. Biz onu kaybetmek istemiyoruz. Allah rızası için sesimizin ulaştığı herkes kendi akraba, komşu ve çevrelerinde Alara’yı tanıtıp bize yardım etsin. Durumu daha iyi olan zenginlerimizden büyük yardımlar istiyoruz. Kampanyamızın bitmesine çok az kaldı. Kızımız ağır sağlık problemleriyle karşılaşmadan acilen tedavi olması gerekiyor.”
























YORUMLAR